Anderson-Fabry disease (AFD), or Fabry disease (FD), was first described by two dermatologists, William Anderson and Johannes Fabry, independently at the end of nineteenth century (1). FD is a lysosomal storage disorder, characterized by functional lack of the enzyme α-galactosidase A (α-Gal A), affecting glycosphingolipid metabolism.
This deficit causes globo-triaosylceramide (Gb3) and lyso-Gb3 storage in lysosomes of several cell types, especially in vascular endothelial cells (2), with consequent multi-systemic effects, affecting renal, cardiac and cerebrovascular systems. These dysfunctions are responsible of significant morbidity and premature death around fourth or fifth decade of life (3).
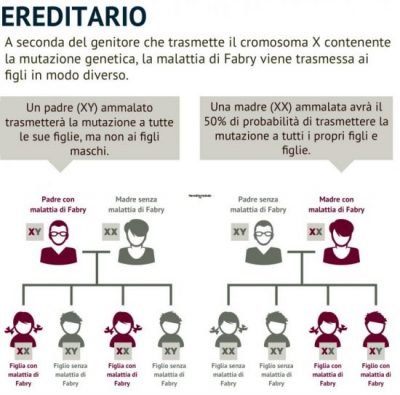
FD is an X-linked lysosomal storage disorder caused by mutations in GLA gene, which encodes for α-Gal A (2). Nowadays, more than 900 different variants in GLA gene are known.Clinical signs, anamnesis and medical history should guide the clinicians to investigate on GLA enzymatic and genetics alteration that causes FD. The partial or complete loss of α-Gal A activity, tested with biochemical assays, and pathogenic variants identification in GLA gene can confirm Fabry disease diagnosis. Furthermore, the determination of Gb3 and lyso-Gb3 storage provides a diagnostic confirm.
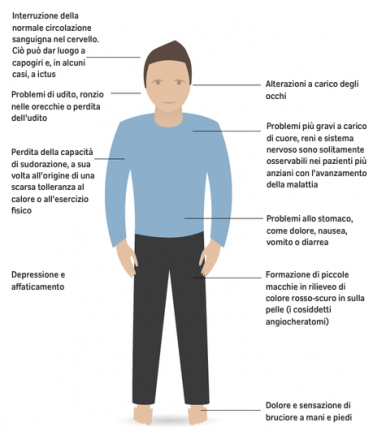

FD onset can occur at different age, generally in childhood, with a broad spectrum of clinical phenotypes (3). The classical signs and symptoms arise already in children with burning acroparesthesia.
The autonomic nervous system impairment is responsible for the alteration of sweating, which becomes a critical point during fever or physical stress: in these conditions, patients can manifest the typical crises of pain, due to the thermoregulation disorder.
Also the gastrointestinal pain, associated with diarrhea and abdominal fullness, is caused by the involvement of the vegetative system alteration. Fabry patients’ skin can manifest typical angiokeratomas, especially in the swimsuit area or as angiokeratomas corporis diffusum and lymphedema in lower limbs. The most common ocular manifestation of FD, as well as others metabolic disorders, is the cornea verticillata; cataracts and conjunctival telangiectasia are less frequent. Glycosphingolipids storage in kidney cells affects their function, leading to progressive nephropathy, one of the most critical manifestation in FD patients. Cardiac involvement is frequent in FD patients in the form of apparently idiopathic left ventricular hypertrophy, alteration of valvular apparatus and intracardiac conduction.
Chronic heart failure is the predominant cause for substantially increased morbidity and reduced life expectancy (4). Moreover, FD patients present a direct involvement of the brain with juvenile ischemic strokes and cerebral bleed.
Besides FD patients with a classical phenotype, an increasing number of individuals with atypical variants and mild phenotype (late-onset and single-organ), has been identified (5, 6). These atypical variants are associated to GLA exonic mutations, and also males can have residual enzymatic activity. Females, usually heterozygous, have chimeric organs containing healthy and affected cells, due to random X-chromosome inactivation. Generally, these women have mild symptoms, therefore it is more difficult to make FD diagnosis (7). Until few years ago, female patients were considered carriers, but this idea was revised and today it is known that female patients can present severe manifestations of the disease.(8). FD was considered a rare disease, but recently literature data indicate it as uncommon and little-known disorder (9). FD diagnosis is still difficult due to disease peculiarities, since clinical signs and symptoms overlap with other pathologies, and it is possible to make several differential diagnoses. The misdiagnosis is a real risk that leads to underestimate the number of individuals affected by FD (10).
Bibliography
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163-7.
- Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw-Hill; 2001:3733-74.
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 22;5:30.
- Gambarin FI, Disabella E, Narula J, et al. When should cardiologists suspect Anderson-Fabry disease? Am J Cardiol. 2010;106(10):1492-9.
- Nakao S, Takenaka T, Maeda M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333(5):288-93.
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64(3):801-7.
- Morey C, Avner P. Genetics and epigenetics of the X chromosome. Ann N Y Acad Sci. 2010;1214:E18-33.
- Pinto LL, Vieira TA, Giugliani R, Schwartz IV. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010;5:14.
- Hoffmann B, Mayatepek E. Fabry disease-often seen, rarely diagnosed. Dtsch Arztebl Int. 2009;106(26);440-7.
- Marchesoni CL, Roa N, Pardal AM, et al. Misdiagnosis in Fabry disease. J Pediatr. 2010;156(5):828-31.
LINK:
Research and Diagnosis Center Malattia di Anderson-Fabry
Monothematic Volume Anderson-Fabry disease on GTND Journal of Nephrological and Dialytic Techniques 2017
Articles:
Genetic screening of Fabry patients with EcoTILLING and HRM technology. 2011 Bono Duro et al.