La malattia di Anderson-Fabry (o Fabry) prende il nome dai due dermatologi che in maniera del tutto indipendente la descrissero per la prima volta alla fine dell’Ottocento (1). Si tratta di un disordine metabolico, da accumulo lisosomiale, caratterizzato dal deficit funzionale dell’enzima α-galattosidasi A (α-Gal A). Tale deficit determina un’alterazione del metabolismo di alcuni glicosfingolipidi, prevalentemente globo-triaosilceramide (Gb3) e lyso-Gb3, che di conseguenza si accumulano nei lisosomi di numerosi tipi cellulari, soprattutto nelle cellule dell’endotelio vascolare (2). Ciò provoca manifestazioni cliniche sistemiche e complicanze di natura renale, cardiaca e cerebrovascolare o una combinazione di esse, che possono portare, intorno alla quarta o quinta decade di vita, a una morte prematura (3). Dal punto di vista eziologico, la Fabry è una enzimopatia lisosomiale X-linked determinata da mutazioni nel gene GLA, che codifica per l’α-Gal A (2). Ad oggi sono state descritte circa 900 mutazioni in pazienti Fabry. Il sospetto della malattia viene avanzato in base ai dati clinici, a quelli anamnestico-familiari e infine confermato attraverso analisi genetiche e biochimiche quali l’individuazione dell’alterazione genica specifica e il dosaggio dell’attività dell’α-galattosidasi A, che può essere nulla o deficitaria. Anche la determinazione dei substrati dell’enzima, Gb3 e lyso-Gb3, fornisce un supporto diagnostico.
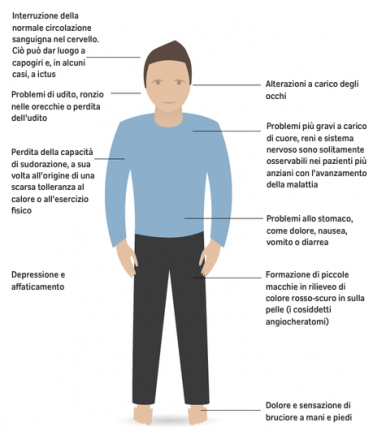

I primi sintomi possono manifestarsi in età diverse, anche se generalmente nel periodo infantile, e avere entità e decorsi differenti (3). Il quadro clinico classico della malattia di Fabry insorge generalmente già in età pediatrica con acroparestesie urenti. La compromissione del sistema nervoso autonomo è responsabile dell’alterazione della sudorazione, che diviene elemento critico in particolari situazioni come la febbre e lo sforzo fisico, momenti in cui, per il disordine della termoregolazione, si possono avere delle tipiche crisi di dolore. Anche le manifestazioni dolorose gastrointestinali, associate a diarrea alternata a senso di pienezza addominale, trovano spiegazione nel coinvolgimento del sistema vegetativo locale. La cute nella malattia di Fabry può manifestare i tipici angiocheratomi, espressi principalmente nelle aree della “mutandina” o, addirittura, come angiocheratoma corporis diffusum e la presenza di un linfedema, soprattutto degli arti inferiori. L’occhio, come in molte malattie neuro-metaboliche, è interessato con espressioni che vanno dalla tipica cornea verticillata, alla cataratta, alle teleangectasie della congiuntiva. L’accumulo di glicosfingolipidi nelle cellule renali ne compromette la funzione, portando a una nefropatia progressiva che è una delle caratteristiche più critiche della malattia. Nei pazienti Fabry il cuore presenta frequentemente un’ipertrofia ventricolare sinistra, apparentemente idiopatica, una diretta compromissione dell’apparato valvolare e di conduzione intracardiaca, con complicanze che vanno dall’insufficienza cardiaca alla patologia aritmica, che può portare a morte per infarto del miocardio (4). Oltre che la compromissione del sistema nervoso periferico, il paziente con malattia di Fabry presenta un coinvolgimento diretto dell’encefalo con stroke ischemici giovanili e possibili episodi emorragici.
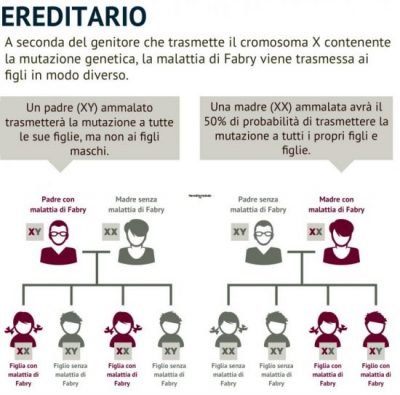
Oltre al fenotipo classico, sono identificati un numero crescente di individui con un fenotipo attenuato, varianti atipiche (5, 6). In letteratura sono riportati numerosi casi di malattia di Fabry, a esordio tardivo e sintomatologia sfumata, che manifestano sintomi a carico di un solo organo. Anche le varianti atipiche sono associate a mutazioni esoniche e un’attività enzimatica residua può essere presente anche nei maschi. Gli individui di sesso femminile, di solito eterozigoti, i cui organi sono chimere di cellule normali e malate, a causa della lyonizzazione del cromosoma X, hanno, generalmente, sintomi meno evidenti e quindi di più difficile individuazione (7). Fino a qualche anno fa si riteneva fossero privi di sintomi ma in realtà anche le femmine possono presentare, nelle fasi tardive, disordini cardio- e cerebrovascolari della stessa gravità degli individui emizigoti (8).
La malattia di Fabry è considerata rara ma, come emerge dalla recente letteratura, sarebbe più giusto considerarla un disordine non comune, poco conosciuto (9). La diagnosi risulta ancora oggi difficile proprio per le peculiarità della malattia, che si presenta con manifestazioni cliniche sovrapponibili a quelle di altre patologie e un’ampia possibilità di diagnosi differenziali che coinvolgono diverse specializzazioni mediche. L’errore diagnostico è un rischio concreto che determina una sottostima del reale numero di soggetti affetti (10).
Bibliografia
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163-7.
- Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw-Hill; 2001:3733-74.
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 22;5:30.
- Gambarin FI, Disabella E, Narula J, et al. When should cardiologists suspect Anderson-Fabry disease? Am J Cardiol. 2010;106(10):1492-9.
- Nakao S, Takenaka T, Maeda M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333(5):288-93.
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64(3):801-7.
- Morey C, Avner P. Genetics and epigenetics of the X chromosome. Ann N Y Acad Sci. 2010;1214:E18-33.
- Pinto LL, Vieira TA, Giugliani R, Schwartz IV. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010;5:14.
- Hoffmann B, Mayatepek E. Fabry disease-often seen, rarely diagnosed. Dtsch Arztebl Int. 2009;106(26);440-7.
- Marchesoni CL, Roa N, Pardal AM, et al. Misdiagnosis in Fabry disease. J Pediatr. 2010;156(5):828-31.
LINK UTILI:
CENTRO RICERCA E DIAGNOSI Malattia di Anderson-Fabry
Volume Monotematico Malattia di Anderson-Fabry su GTND Giornale Tecniche Nefrologiche e Dialitiche 2017
Articoli:
Genetic screening of Fabry patients with EcoTILLING and HRM technology. 2011 Bono Duro et al.