Dal 2021 presso il Centro di Diagnosi e Ricerca di Malattie da Accumulo Lisosomiale dell’Istituto per la Ricerca e l’Innovazione Biomedica del CNR di Palermo, ci occupiamo anche dello studio delle alterazioni enzimatiche e genetiche della malattia di Niemann Pick di tipo A (ORPHA 77292) e B (ORPHA 77293).
I tipi A e B della malattia di Niemann Pick o Deficit da Sfingomielinasi acida (ASMD) possono essere considerati gli estremi di un continuum patologico caratterizzato dalla medesima alterazione genica che viene trasmessa con modalità autosomica recessiva. In entrambi i tipi, infatti è coinvolto il gene SMPD1 (sphingomyelinphosphodiesterase 1), localizzato sul braccio corto del cromosoma 11 (locus 11p15.4). Oltre un centinaio di mutazioni, puntiformi, piccole delezioni, alterazioni di splicing, sono descritte in letteratura [1-3]. Questo gene codifica per la sfingomielinasi acida (ASM), un enzima di 631 amminoacidi che presiede alla degradazione della sfingomielina in ceramide e fosforilcolina, il cui mancato o deficitario funzionamento comporterà un alterato metabolismo della sfingomielina che si accumulerà all’interno di diversi tipi cellulari, dando origine al meccanismo che sta alla base della malattia.La malattia è panetnica e la sua incidenza nella popolazione generale varia da 1:100.000 a 1:264.000. Il tipo A è più diffuso tra gli ebrei Ashkenazi, mentre il tipo B presenta percentuali di incidenza più elevate nelle popolazioni del Nord Africa, Turchia e Paesi Arabi[1,4-5].Dal punto di vista clinico, il tipo A, caratterizzato da esordio precoce, in epoca neonatale, presenta un quadro clinico definito da epatosplenomegalia, arresto del normale sviluppo psicomotorio, progressiva perdita di tono muscolare per giungere ad una franca ipotonia e deterioramento neuromotorio sempre più evidente[6]. Una elevata percentuale di pazienti all’esame del fondo oculare presenta una macchia rosso ciliegia in sede maculare. La morte, non raramente per insufficienza respiratoria, avviene nei primi anni di vita.
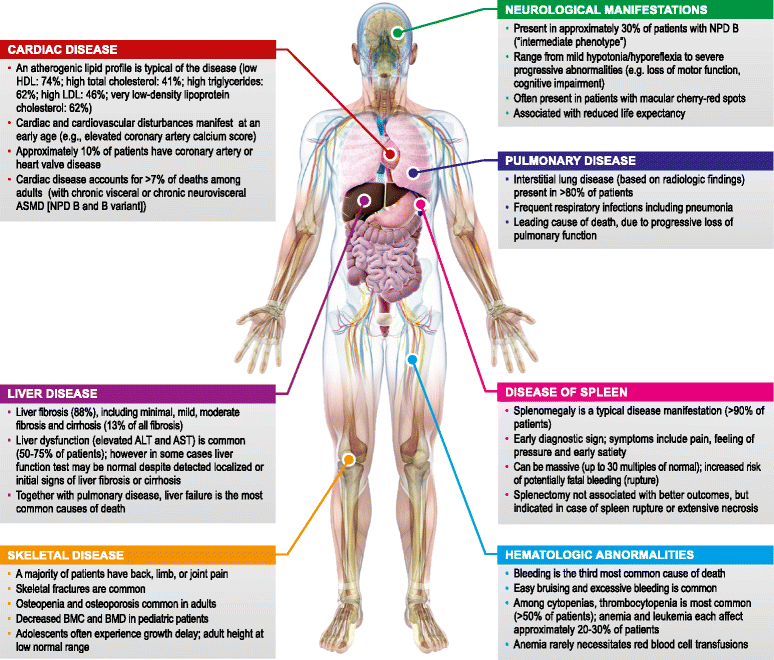
Anche per il Deficit da Sfingomielinasi acida sono stati documentati ritardi diagnostici e casi di misdiagnosi soprattutto con la malattia di Gaucher che per alcuni aspetti presenta un quadro clinico sovrapponibile alla malattia di Niemann Pick di tipo A e B. La diagnosi della malattia può essere eseguita tramite dosaggio dell’attività dell’enzima sfingomielinasi acida. L’indagine genetica è fondamentale per la conferma della malattia, per la prognosi e per la consulenza genetica. Per quanto riguarda il trattamento, l’ASMD è una malattia genetica progressiva e potenzialmente fatale con nessuna opzione di trattamento approvata, al momento. I risultati degli studi clinici condotti su un nuovo farmaco, l’olipudasi alfa, su pazienti adulti e pediatrici, dimostrano chiaramente il potenziale di questo farmaco sperimentale di agire sulle manifestazioni cliniche dell’ASMD e di avere un impatto significativo sulla vita di chi è affetto da questa patologia devastante.
La malattia di Niemann Pick tipo B definita come forma non neuropatica, presenta un decorso ad andamento cronico, composto da epatosplenomegalia, ritardo o perturbamento dell’accrescimento staturale e compromissione polmonare. Sono state descritte anche alterazioni emato-chimiche quali trombocitopenia, leucopenia, sintomatologia algica ossea e osteopenia con ritardo nella maturazione scheletrica. Meno frequente, rispetto al tipo A, è il riscontro della macchia rosso ciliegia in sede maculare [7]. Sono stati descritti, in letteratura, pazienti con caratteristiche cliniche intermedie rispetto ai tipi A e B sopracitati. Essendo una patologia autosomica recessiva, affinché il fenotipo clinico si manifesti, è necessario che entrambe le copie alleliche (quella di origine materna e quella di origina paterna) siano mutate, pertanto il paziente affetto dalla malattia di Niemann Pick di tipo A e B può essere omozigote per una singola mutazione o eterozigote composto (due mutazioni diverse, una nell’allele trasmesso dal padre ed una in quello trasmesso dalla madre). I genitori (geneticamente definiti eterozigoti) solitamente hanno una sola mutazione e non presentano espressione clinica della malattia: sono i cosiddetti portatori sani. Due genitori eterozigoti hanno il 25% di probabilità di avere un figlio affetto dalla malattia e, non essendo una patologia legata ai cromosomi sessuali, entrambi i sessi sono ugualmente colpiti.

Bibliografia
-
- Simonario CM et al., The Demographics and Distribution of Type B Niemann-Pick Disease: Novel Mutations Lead to New Genotype/Phenotype Correlations Am J Hum Genet 2002; 71(6): 1413–1419.
- Rodriguez-Pascau, L et al., Identification and characterization of SMPD1 mutations causing Niemann-Pick types A and B in Spanish patients. Hum. Mutat 2009; 30:1117-1122.
- Zampieri S et al., SMPD1 mutation update: database and comprehensive analysis of published and novel variants, Hum Mutat 2016; 37:139–147>
- Meikle PJ et al., Prevalence of Lysosomal Storage Disorder. JAMA 1999; 281:249-254.
- Schuchman EH The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. 2007; 30(5):654-63.
- McGovern MM et al., Natural history of type A Niemann-Pick disease; possible endpoints for therapeutic trials, Neurology 2006; 66:228–232.
- Schuchman EH et al., Types A and B Niemann-Pick disease Mol. Genet. Metab 2017; 120:27-33.