Il Centro di Ricerca e Diagnosi Malattie da Accumulo Lisosomiale sito presso l’Istituto di Biomedicina ed Immunologia Molecolare del CNR si occupa anche dello studio delle alterazioni enzimatiche e genetiche che causano la Malattia di Pompe o Glicogenosi di tipo II (GSDII) o deficit della maltasi acida (AMD). Si tratta di una patologia neuromuscolare rara, cronica, debilitante e spesso mortale. La malattia di Pompe, che presenta diverse manifestazioni cliniche che variano a seconda dell’età di insorgenza, può essere classificata in due forme: infantile (Infantile Onset Pompe Desease, IOPD) e tardiva (Late Onset Pompe Desease, LOPD), quest’ultima si può presentare in età giovanile o adulta. La forma infantile si manifesta alla nascita o entro i primissimi mesi di vita, è caratterizzata da cardiomiopatia e ipotonia muscolare (floppy baby o bambole di pezza) ed è più grave rispetto alla LOPD. La IOPD se non riconosciuta e trattata in maniera tempestiva, può causare la morte nel primo anno di vita. La forma ad esordio tardivo mostra un ampio spettro di fenotipi clinici con diverse velocità di progressione e con coinvolgimento della muscolatura prossimale e respiratoria.
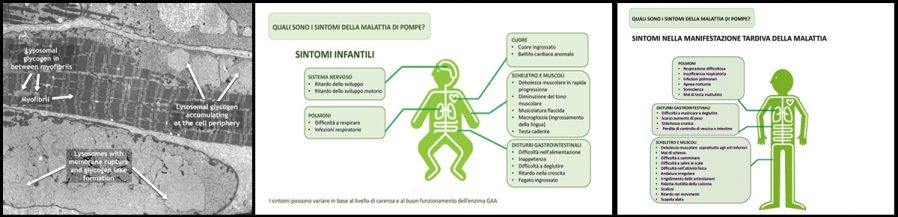
La Malattia di Pompe è un disordine autosomico recessivo del metabolismo del glicogeno causata dalla mancanza o dal deficit dell’enzima alfa-1,4-glucosidasi acida (GAA, maltasi acida), deputato allo smaltimento del glicogeno nei lisosomi. I genitori portatori sani di una mutazione spesso non sanno di averla. Essendo una patologia autosomica recessiva, se entrambi i genitori sono portatori sani, ad ogni gravidanza avranno un rischio del 25% di generare figli affetti, il 50% di probabilità di avere figli portatori sani e il 25% di avere figli sani non portatori. Al momento sono note più di 450 mutazioni diverse sul gene che codifica per alpha-1,4-glucosidasi acida. Perché la malattia si manifesti come parziale o completa perdita di attività della GAA, entrambe le copie del gene GAA devono avere una mutazione di sequenza considerata patogena. Alcune mutazioni sono ampiamente diffuse tra i pazienti con malattia di Pompe (per esempio c.-32-13T>G, nella forma ad esordio tardivo è frequentemente riscontrata in eterozigosi con un’altra mutazione sul secondo allele), alcune mutazioni sono “private”, trovate solo in una singola famiglia o in una piccola popolazione. La malattia di Pompe, all’insorgenza, tende ad essere ignorata: si inizia con l’esclusione delle malattie più comuni, ma questo porta a ritardi della diagnosi. I sintomi clinici (singolarmente o in qualsiasi combinazione) come la presenza di debolezza muscolare prossimale progressiva inspiegabile, con o senza sintomi respiratori e/o l’aumento della creatina chinasi (iperckemia) asintomatica dovrebbero indirizzare i clinici ad approfondire lo studio di eventuali alterazioni enzimatiche e genetiche di GAA che possono causare la LOPD.
Nei neonati, una diagnosi precoce è molto importante in quanto, senza trattamento, la morte avviene entro il primo anno di vita. Un’analisi dei dati del registro Pompe ha evidenziato ritardi della diagnosi sia in neonati e bambini, che negli adolescenti e negli adulti. La tempestività della diagnosi è un fattore cruciale per questa patologia, esistono varie tipologie di test diagnostici per la sua rilevazione, il più comune è un test enzimatico, volto alla misurazione dell’attività enzimatica del GAA, eseguito attraverso il prelievo di sangue (DBS, Dried Blood Spot). Brevemente, una goccia di sangue viene adsorbita su carta da filtro e lasciata asciugare a temperatura ambiente. L’attività enzimatica viene saggiata con un metodo fluorimetrico. L’analisi genetica è una soluzione per diagnosticare l’effettiva presenza di mutazioni, nella sequenza del gene GAA, responsabili dell’insorgenza della patologia e per rilevare i portatori sani nel quadro familiare, che potenzialmente possono trasmetterla ai figli.

Dal 2016 esiste una terapia enzimatica sostitutiva (ERT) per la malattia di Pompe basata sull’utilizzo della alfa-glucosidasi acida ricombinante umana. Il nostro gruppo di ricerca utilizza metodiche enzimatiche e biomolecolari necessarie a diagnosticare la malattia e con la collaborazione dei clinici coinvolti si può agevolare il percorso diagnostico nei pazienti che hanno ricevuto un sospetto clinico di essere affetti da questa patologia rara.